File formats
Input
The input contact map is stored as a hash structure using pair-bins as keys. Then, a query is binned into a corresponding key based on its position. The size of the bin is specified by a user (-in_hic_resol for .hic) or dependents on the input file (bin-contact pair files).
HiC map
a bin-contact pair format
-in_bin
A bin file defines the chromosome, start and end positions of each bin, an example: fly_30k.cbins (D.mel in 30k bin).
cbin chr from.coord to.coord
1 2L 6000 7000
2 2L 7000 8000
3 2L 8000 9000
4 2L 9000 10000
5 2L 12000 13000
-in_map
A contact map contains Hi-C frequency indexed by bins in text format, an example fly_30k.n_contact.
cbin1 cbin2 expected_count observed_count
1 1 0.077080 50
1 2 0.389912 314
1 3 0.493750 163
1 4 0.560505 169
1 5 0.368884 79
expected_count : the expected contact between those two genome locis (bins) according to model. It could be 1 if no model is applied.
observed_count : the observed contact between those two genome locis (bins) in Hi-C data
It takes time to parse text format so we develop bin/genBinMap to turn text format (.n_contact) into binary format (.binmap) which could be used in -in_map, an example fly_30k.binmap.
genBinMap [options] -in_ncontact input.n_contact -out_binmap out.binmap
>bin/genBinMap -in_ncontact examples/fly_30k.n_contact -out_binmap examples/fly_30k.binmap
>hicmaptools -in_map examples/fly_30k.binmap -in_bin examples/fly_30k.cbins -bait examples/bait.bed -output baitTest.tsv
.hic format
The . hic format is generated by Juicer. The parser of . hic is adapted from straw.
-in_hic
A .hic file by applying Juicer on HiC fastq of Galaxy training.
-in_hic_norm (optional)
To extract data based on a specified normalization method, NONE, VC, VC_SQRT, or KR (default: NONE).
-in_hic_resol (optional)
To extract data at a specified resolution, e.g., 5000, 10000, or 50000 (default: 10000).
Query file
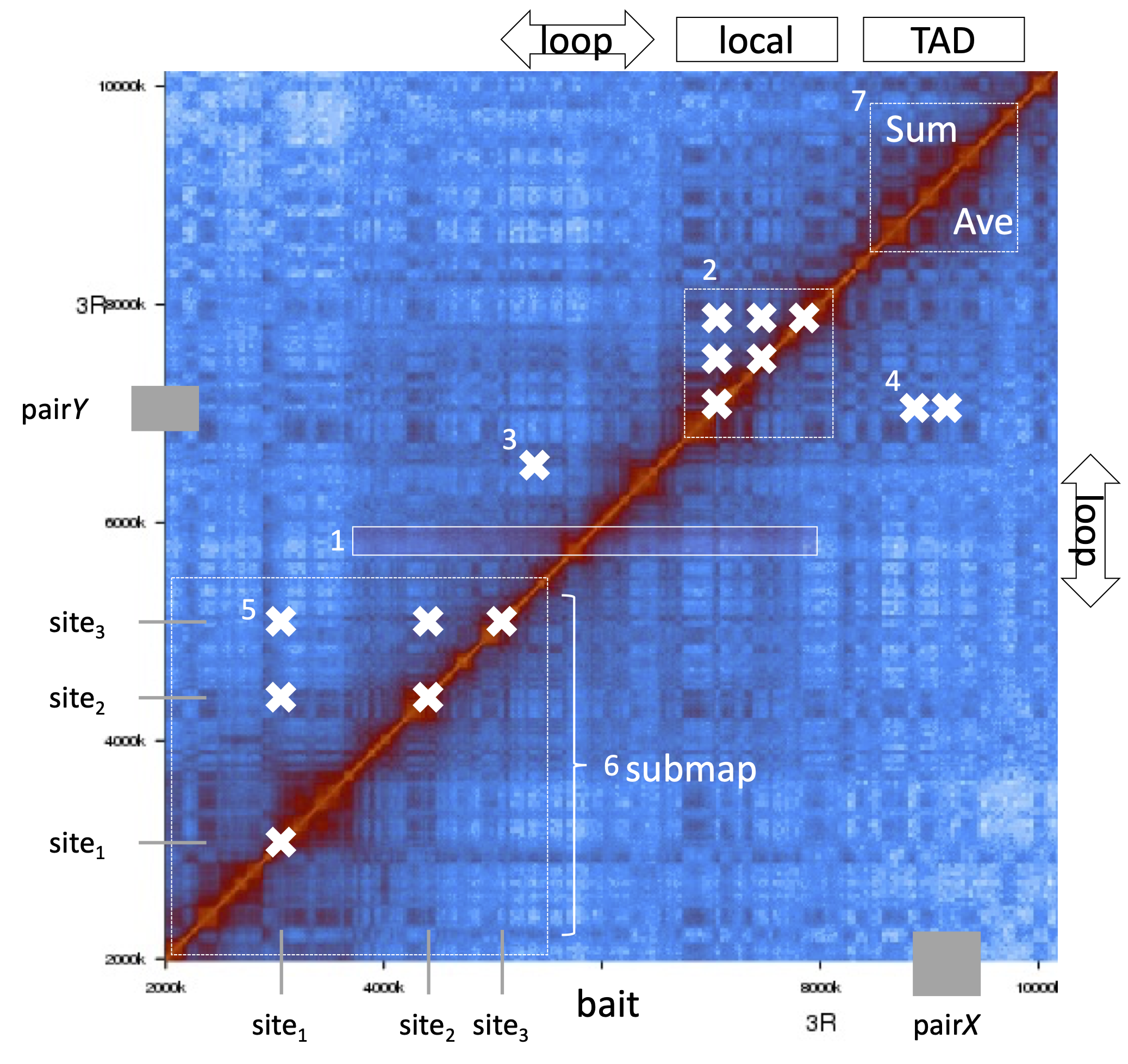
The query file is in bed format, where the first three columns are enough. Next, the example of each query mode is listed. Although the biology scenarios of examples are mainly based on Drosophila, HiCmapTools could handle other species (i.e., the example of the mouse Tox gene in the bait query model).
bait: bait.bed a PRE binding site, Tox_mm10.bed mouse Tox gene
local: local.bed a PcG TAD
loop: loop.bed gene, Antp
pair: pair.bed a pair of insulator binding sites
sites: sites.bed a list of insulator binding sites in range 3L:10000000-11000000
submap: submap.bed a region contains Antp-BX long range conttact
TAD: TAD.bed selected TADs in range 2L:2000000~3000000
Illustration of query modes
Output
There are two output files. You can use the tool tools/visualPermutationTest.R to examine query’s contact frequency aganist the null hypothesis (Shuffle test).
output.tsv : the contact frequency of the interested regions
sum_* indicates frequency of HiC
rand_* indicates frequency of shuffle test
divide_* indicates ratio of sum/rand
rank_* indicates the rank of HiC among shuffle test. The smaller rank is, the stronger query frequency is (i.e., rank_nor 0.600 = top60%).
index chrom start end sum_obs sum_exp sum_nor rand_obs rand_exp rand_nor divide_obs divide_exp divide_nor rank_obs rank_exp rank_nor
1 2L 594629 595145 47916.000 459.715 2380.531 32618.180 314.679 2525.479 1.469 1.461 0.943 0.100 0.140 0.600
output _random .txt : the observed, expected and normalizated contact intensities of the null hypothesis starting from the third row where the second row is the query frequency
random_obs,random_exp,random_nor
47916,459.715,2380.53
19632,158.539,2956.25
57574,448.25,2832.44
7074,60.7897,3029.22
33009,246.588,3311.8